环氧树脂/聚砜共混体系相结构的调控研究
――环氧预聚物分子量的影响
摘 要:研究了不同分子量的环氧预聚物对双酚A型双官能团环氧树脂/聚砜(PSF)/固化剂(二氨基二苯基砜,DDS)体系相分离结构的影响.通过红外光谱(FTIR)和动态热机械分析(TMA)对反应转化率、玻璃化温度以及固化温度的关系的研究,表明环氧预聚物分子量较小时,凝胶点和玻璃化是影响相结构的关键因素;环氧分子量较大时,环氧扩链后粘度的变化则成为抑制相分离的重要因素.电子显微镜(SEM)结果表明改变环氧预聚物分子量可以达到调控相结构的目的,随着预聚物分子量的增大,体系的微区尺寸减小.
关键词:环氧树脂,共混物,相结构,粘度,分子量
BLENDS BY MOLECULAR WEIGHT OF EPOXY RESINS
(Laboratory of Advanced Materials Institute of Polymer Science and Engineering,
Department of Chemical Engineering, Tsinghua University,Beijing 100084)
Abstract:Phase separation during the
curing reaction in blends of diglycidly ether of bisphenol A,polysulfone
and diaminodiphenylsulfone(DDS,curing agent) was studied. Effects of epoxy
resins with different molecular weight on phase structure of the blends
were investigated. The relation between the degree of conversion and
reaction time revealed that vitrification and gelation greatly affected
the formation of phase structure in the low molecular weight epxoy
systems. When Tg of the epoxy-rich phase went up to the
reaction temperature, both reaction and phase separation were suppressed.
While in the high molecular weight epoxy systems, the viscosity increasing
of epoxy resins played an important role. Although the degree of
conversion of the epoxy-rich phase did not reach the gel point after 24h
precure, phase separation was suppressed due to the high viscosity of the
systems. SEM results showed that the desired phase structure could be
achieved by using epoxy resins with different molecular
weight.
Key words:Epoxy
resin, PSF, Viscosity, Molecular weight
反应诱导相分离是指在单体和聚合物混合体系中,由于单体的聚合反应导致混合物中组分变化并引发不相容所产生的相分离现象.用反应来诱导相结构的形成是多组分聚合物领域的新课题[1,2].环氧树脂/热塑性树脂共混体系是典型的反应诱导相分离体系.在这种体系中相结构的形成是热力学和动力学综合作用的结果.热力学方面,环氧低聚物进行扩链反应,分子量不断增大,环氧树脂与热塑性树脂的相容性下降,促进了相分离的发展;动力学方面,在固化反应后期,随着交联网络的形成,体系粘度无限增大,热塑性树脂分子链的扩散运动受到限制,相分离受到抑制.因此,调节固化反应和相分离速度的相对快慢,可以达到调控相结构的目的.近年来,一些学者沿着这一思路研究了含环氧的共混体系的相分离过程,尝试着进行相结构的控制[2~8].
Inoue等通过研究认为[1],环氧/热塑性树脂体系的相结构发展通常遵循旋节相分离(Spinodal
decomposition,SD)的机理,相区连续性和相区尺寸均随时间变化.固化反应初期,均相混合物发生相分离,产生双连续结构.随着相分离的进行,形成周期间距(Periodic
distance)相似的结构.同时由于界面能的增加,相连续性被破坏,形成球粒结构.由于体系流动性降低,粒子在原处粗化,最终形成彼此相连的球粒结构.如果能将相分离过程在不同的发展阶段固定住,就可以得到不同的相结构.这种观点得到了许多学者的认可[3~8].
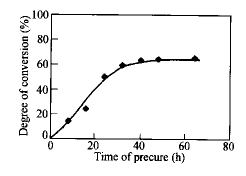
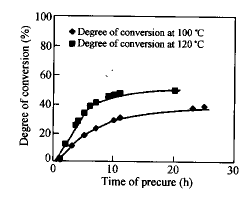
Hsich等研究了橡胶改性的环氧树脂体系的相分离结构,结果表明通过选择固化反应温度和活性不同的固化剂可得到不同的相结构[5].本课题组前一阶段的研究表明[6~8],在较低的温度下预固化,调控预固化时间,然后升温后固化,可以达到调控相结构的目的.由于共混体系的固化反应和相分离速度与环氧树脂的初始分子量有依赖关系,因此本研究直接选用不同分子量的环氧预聚物,探讨环氧初始分子量对共混物相结构的影响.
1 实验部分
1.1 实验原料及固化样品的制备
不同分子量环氧树脂预聚物,E56(MW=357),E51(MW=392),E42(MW=476),E39(MW=513),E31(MW=695),巴陵石化公司产品.聚砜(PSF)(特性粘度0.35)由吉林大学提供.固化剂二氨基二苯基砜(DDS),共溶剂二氯甲烷.
环氧树脂与聚砜的组成比为100/30(重量比),使用与环氧等当量的固化剂,将加入共溶剂的环氧树脂、聚砜和固化剂的混合物,在100℃油浴中搅拌均匀.将样品倒入一开放模具中抽真空,去除共溶剂,按一定程序固化.
1.2 样品的观测
将混合均匀的样品涂在KBr单晶片上,在红外灯下去除溶剂后用岛津FTIR-8201PC原位测定固化反应动力学.以环氧基在916cm-1处特征峰的消耗率来表征反应程度,选1148cm-1处S![]() O基的特征峰为基准峰.反应程度按下式计算:
O基的特征峰为基准峰.反应程度按下式计算:
其中E0和Et分别是环氧基特征峰在反应前和反应到t时刻与基准峰的吸收强度比,Xt为t时刻的反应程度.
将固化后的样品在液氮温度下脆断,二氯甲烷刻蚀断面,干燥后镀金膜,在日立S-450电镜上观测相结构.
将混合后的样品均匀地涂在玻璃丝上,去除溶剂,固化后在扭辫式动态力学测试仪(GDP-Ⅱ型(Torsional
braid analysis,TBA)上测试玻璃化温度(Tg).
2 结果与讨论
2.1 改变环氧预聚物分子量对环氧/聚砜/二氨基二苯基砜体系相结构的调控
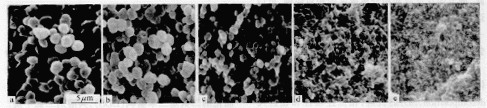
选用五种不同分子量的环氧预聚物(分别为E56、E51、E42、E39、E31)分别与聚砜共混,在100℃预固化4h后在160℃后固化,最终的相结构如图1所示.从图中可以看出,环氧预聚物分子量不同,按同样的程序固化后,得到的相结构不同.随着预聚物分子量的增大,环氧富集相微区尺寸减小.我们认为相结构的不同是由于初始分子量的不同,导致共混体系粘度不同.